

NIH researchers identify key genomic features that could differentiate SARS-CoV-2 from other coronaviruses that cause less severe disease

The Pandemic Is Propelling a New Wave of Automation
15/06/2020
Researchers tap location data to capture effect of Covid-19 policies
15/06/2020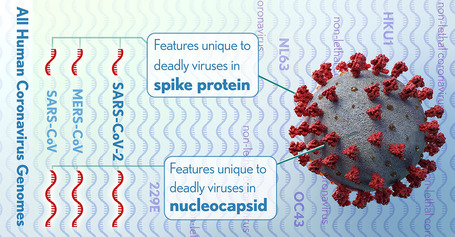
Research could be a crucial step in helping scientists develop approaches to predict severity of future coronavirus disease outbreaks.
A team of researchers from the National Library of Medicine (NLM), part of the National Institutes of Health, identified genomic features of SARS-CoV-2, the virus that causes COVID-19, and other high-fatality coronaviruses that distinguish them from other members of the coronavirus family. This research could be a crucial step in helping scientists develop approaches to predict, by genome analysis alone, the severity of future coronavirus disease outbreaks and detect animal coronaviruses that have the potential to infect humans. The findings were published this week in the Proceedings of the National Academy of Sciences(link is external).
COVID-19, an unprecedented public health emergency, has now claimed more than 380,000 lives worldwide. This crisis prompts an urgent need to understand the evolutionary history and genomic features that contribute to the rampant spread of SARS-CoV-2. “In this work, we set out to identify genomic features unique to those coronaviruses that cause severe disease in humans,” said Dr. Eugene Koonin, an NIH Distinguished Investigator in the intramural research program of NLM’s National Center for Biotechnology Information, and the lead author of the study. “We were able to identify several features that are not found in less virulent coronaviruses and that could be relevant for pathogenicity in humans. The actual demonstration of the relevance of these findings will come from direct experiments that are currently getting under way.”
Using integrated comparative genomics and machine learning techniques, the researchers compared the genome of the SARS-CoV-2 virus against the genomes of other members of the coronavirus family and identified protein features that are unique to SARS-CoV-2 and two other coronavirus strains with high fatality rates, SARS-CoV and MERS-CoV. The identified features correspond with the high fatality rate of these coronaviruses, as well as their ability to move from animal to human hosts.
These features include insertions of specific stretches of amino acids into two virus proteins, the nucleocapsid and the spike.
These features are found in all three high-fatality coronaviruses and their closest relatives that infect animals, such as bats, but not in four other human coronaviruses that cause non-fatal disease. In particular, the insertions in the spike protein are predicted, from protein structure analysis, to facilitate the recognition of the coronavirus receptors on human cells and the subsequent penetration of the virus into those cells. Finding these features in animal coronavirus isolates could predict the jump to humans and the severity of disease caused by such isolates.
Source: www.nih.gov
